EpDis and MassPred Documentation
Table of Contents
I. MassPred
MassPred is a set of tools that provides easy predictor installation, application of predictors to input data and filtering of the results of predictor action. MassPred takes the contents of input files or directories, extracts every single protein and applies the desired predictors to the extracted proteins, creating separate jobs that can be simultaneously executed on a symmetric multiprocessor computer, or on computers with a multicore/multithread processor architecture. MassPred itself does not perform a parallel execution of a single predictor application to a single protein. MassPred tool is developed for Linux operating system, and optimized for Ubuntu Linux version.
Current version is 0.38. Download link: MassPred.0.38.tar.gz
1.1 Description
MassPred is a command line (shell) oriented system which enables multiple (massive) and parallel execution of predictors with group FASTA files. Results are filtered and stored in a form suitable for loading into SQL. Also MassPred accepts multiple proteins in the FASTA file. Also, it can be used to execute any command on a set of proteins.
MassPred is a set of tools that provides easy predictors installation, application of predictors to input data and filtering of the results of predictor action. MassPred provides scripts for automated installation of the desired predictors and their preparation for automatic execution. Predictors (any of the previously mentioned three types) or methods for hydropathy calculation are applied to the protein dataset, which can be stored in one or more files or directories. Each file can include one or more proteins in FASTA format. MassPred takes the contents of input files or directories, extracts every single protein and applies the desired predictors to the extracted proteins, creating separate jobs for every pair (protein, predictor). The created jobs can be simultaneously executed on a symmetric multiprocessor computer, or on computers with a multicore/multithread processor architecture. MassPred itself does not perform a parallel execution of a single predictor application to a single protein.
MassPred collects the results and filters them in a CSV file format in order to prepare the results in a form that can be used as an input in a LOAD utility program for loading results in RDBMS tables. By default, the results are filtered for IBM DB2 RDBMS, but can be modified for any other relational database system.
The MassPred mechanism for generating jobs for simultaneous execution is not restricted to supported predictors. In fact, any program that takes a single protein in FASTA format can be massively applied to a set of proteins.
1.1.1 List of supported predictors
1.1.1.1 Disorder predictors
- VSL2b
- IUPred (Long, Short) v1.0
- DisEMBL (Coin coils, Hot loops, Remark465) v1.4
- RONN v3.1
- Predisorder v1.1
- IsUnstruct v2.02
- Disopred v2.43
- OnD-CRF v1.0
1.1.1.2 MHC binding predictors
- NetMHCpan 2.0 v2.0c
- NetMHCpan 2.4 v2.4a
- NetMHCpan 2.8 v2.8a
- NetMHCIIpan 1.0 v1.0b
- NetMHCIIpan 2.0 v2.0b
- NetMHCIIpan 3.0 v3.0c
- NetMHC 3.0 v3.0c
- NetMHC 3.4 v3.4a
- NetMHCII 2.2
- IEDB Tool MHC I (ann, smm, smmpmbec)
- IEDB Tool MHC II (nn_align, smm_align)
1.1.1.3 Hydropathy calculation
Hydropathy of peptides or protein regions is calculated as the sum of hydropathy values of all the AAs, according to chosen scale, divided by the number of residues in peptide (protein region). Methods are based on the following two scales:
1.1.1.4 Disorder-binding predictors
1.2 Installation
Package installation is done in two steps: MassPred installation and installation of predictors that are necessary for work.
1.2.1 MassPred installation
Unpack MassPred distribution file with command:
tar xzf masspredx.xx.tar.gz.
Name of directory which includes unpacked version should includes only letters, digits, minus, underline,
point, or commas. Other symbols in the path may cause a problem in MassPred's work.
Recommended directory for installation is /usr/local/masspred.
MassPred directory after unpacking contains sub directory named predictors. This directory
includes sub directory source with the following structure:
clean.sh- shell script which removes all installed predictors;data- directory with source of predictors, downloaded from predictors URL;data.md5- MD5 sum of predictors sources files;download- directory with shell scripts for automatic download (it is available) of predictor source file;download_all.sh- shell script which downloads all available predictors sources files;install- directory with shell scripts for installing predictors;install_all.sh- shell script which installs all predictors;md5sum_data.sh- shell script which calculates MD5 sums of predictors sources files, output must be the same as data.md5;patch- directory with patch files for some predictors;test- directory with shell scripts for testing predictors;test_all.sh- shell script which tests all predictors
Predictors sources files are not included because of the licenses, but some predictors or some of their components are on permanent URL (listed above in list of supported predictors) which is used by download scripts.
1.2.2 Predictors installation
First step in predictor installation process is install the tools necessary for the installation process and operation of predictors. Tools includes specific version of Python and runtime C/C++ libraries. For Ubuntu Linux use the following command:
sudo apt-get install build-essential gawk python-dev tcsh defaultjre libstdc++5 libstdc++5:i386 libstdc++6:i386 libc6:i386
libncurses5:i386 lib32stdc++6 lib32ncurses5 lib32z1 lib32bz21.0
Instruction for the predictors installation are:
- Open terminal window and go to the
predictorsdirectory; - Execute
source/download_all.shto download all available predictors; - If some of predictors requires additional files, download it as described in its own installation requirements;
- Optionally execute
source/md5sum_data.sh > source/data_new.md5to calculate MD5 sums; - Optionally compare
source/data.md5andsource/data_new.md5; generally they should be equal; - Execute
source/install_all.shto install all predictors; - Optionally execute
source/test_all.shto test all predictor(s) installation;
The specific information about download individual predictors and their test after installation are listed in the rest of this chapter.
| ANCHOR-1.0 |
|
|||
| DisEMBL-1.4 |
|
|||
| Disopred-2.43 |
|
|||
| IsUnstruct-2.02 |
|
|||
| IUPred-1.0 |
|
|||
| netMHC-3.0c |
|
|||
| netMHC-3.4a |
|
|||
| netMHCII-2.2 |
|
|||
| netMHCIIpan-1.0b |
|
|||
| netMHCIIpan-2.0b |
|
|||
| netMHCIIpan-3.0c |
|
|||
| netMHCpan-2.0c |
|
|||
| netMHCpan-2.4a |
|
|||
| netMHCpan-2.8a |
|
|||
| OnD-1.0 |
|
|||
| Predisorder-1.1 |
|
|||
| RONN-3.1 |
|
|||
| VSL2b |
|
1.3 Configuration
Before running MassPred, it is necessary to create a configuration file with lines in the following form:
variable=value
Comments are from # to the end of the line.
File configuration.ish in installed directory is an example of configuration file.
Variables that can be put into the configuratio file and their meaning are listed in the table below.
| Name | Description |
|---|---|
CPU_NUMBER |
Specify maximal number of simultaneously executed jobs. Parameter value is number,
with default 4. |
WORK_SQL |
Specify if SQL files with insert statements should be generated. Parameter value is string
with default value "no". Possible values are "yes" or "no". |
WORK_NUMERIC |
Specify generation of alternative form results with numeric value for each AA. Parameter
value is string with default value "no". Possible values are "yes"; or "no". |
WORK_COMMAND |
Specify execution of some command for each protein. Parameter
value is string with default value "no". Possible values are "yes"; or "no" |
COMMAND |
This variable has meaning only if WORK_COMMAND is set. Specify command which
executes for each protein. Parameter value is string with default value "-" (when report
error, because "-" is nonexistent command). |
WORK_HYDRO |
Specify generation of table hydro for each AA in protein. Parameter value is string with default value "no". Possible values are "yes"; or "no" |
WORK_ANCHOR |
Specify execution of ANCHOR predictor. Parameter value is string with default value "no". Possible values are "yes"; or "no" |
WORK_DISEMBL |
Specify execution of DisEMBL. Parameter value is string with default value "no". Possible values are "yes"; or "no" |
WORK_DISOPRED |
Specify execution of disopred predictor. Parameter value is string with default value "no". Possible values are "yes"; or "no" |
WORK_ISUNSTRUCT |
Specify execution of IsUnstruct predictor. Parameter value is string with default value "no". Possible values are "yes"; or "no" |
WORK_IUPRED_LONG |
Specify execution of iupred predictor, long variant. Parameter value is string with default value "no". Possible values are "yes"; or "no" |
WORK_IUPRED_SHORT |
Specify execution of iupred predictor, short variant. Parameter value is string with default value "no". Possible values are "yes"; or "no" |
WORK_OND |
Specify execution of OnD predictor. Parameter value is string with default value "no". Possible values are "yes"; or "no" |
WORK_PREDISORDER |
Specify execution of predisorder predictor. Parameter value is string with default value "no". Possible values are "yes"; or "no" |
WORK_RONN |
Specify execution of RONN predictor. Parameter value is string with default value "no". Possible values are "yes"; or "no" |
WORK_VSL2 |
Specify execution of VSL2 predictor. Parameter value is string with default value "no". Possible values are "yes"; or "no" |
WORK_NETMHC_1_30C |
Specify execution of netMHC predictor, version 3.0c. Parameter value is string with default value "no". Possible values are "yes"; or "no" |
NETMHC_1_30C_ALLELE_FILE |
This variable has meaning only if WORK_NETMHC_1_30C is set. Specifies file with allele.
Path is specified absolute or relative with regard to configuration file. Parameter value is
string with default value "NetMhc.3.0c.pseudo".
|
NETMHC_1_30C_LENGTH_FROM |
This variable has meaning only if WORK_NETMHC_1_30C is set. Specifies from which
length of peptide predictor works. Parameter value is number, with default 8. |
NETMHC_1_30C_LENGTH_TO |
This variable has meaning only if WORK_NETMHC_1_30C is set. Specifies to which length
of peptide predictor works. Parameter value is number, with default 11. |
NETMHC_1_34A_ALLELE_FILE |
This variable has meaning only if WORK_NETMHC_1_34A is set. Specifies file with allele.
Path is specified absolute or relative with regard to configuration file. Parameter value is
string with default value "NetMhc.3.4a.pseudo"
|
NETMHC_1_34A_LENGTH_FROM |
This variable has meaning only if WORK_NETMHC_1_34A is set. Specifies from which
length of peptide predictor works. Parameter value is number, with default 8. |
NETMHC_1_34A_LENGTH_TO |
This variable has meaning only if WORK_NETMHC_1_34A is set. Specifies to which length
of peptide predictor works. Parameter value is number, with default 11. |
WORK_NETMHC_2_22 |
Specifies execution of netMHCII predictor, version 2.2. Parameter value is string with default value "no". Possible values are "yes"; or "no" |
NETMHC_2_22_ALLELE_FILE |
This variable has meaning only if WORK_NETMHC_2_22 is set. Specifies file with allele.
Path is specified absolute or relative with regard to configuration file. Parameter value is
string with default value "NetMhcII.2.2.pseudo".
|
NETMHC_2_22_LENGTH_FROM |
This variable has meaning only if WORK_NETMHC_2_22 is set. Specifies from which length
of peptide predictor works. Parameter value is number, with default 9. |
NETMHC_2_22_LENGTH_TO |
This variable has meaning only if WORK_NETMHC_2_22 is set. Specifies to which length of
peptide predictor works. Parameter value is number, with default 15. |
WORK_NETMHC_PAN_1_20C |
Specifies execution of netMHCPan predictor, version 2.0c. Parameter value is string with default value "no". Possible values are "yes"; or "no" |
NETMHCPAN_1_20C_ALLELE_FILE |
This variable has meaning only if WORK_NETMHC_PAN_1_20C is set. Specifies file with
allele. Path is specified absolute or relative with regard to configuration file. Parameter
value is string with default value "NetMhcPan.2.0c.pseudo".
|
NETMHCPAN_1_20C_LENGTH_FROM |
This variable has meaning only if WORK_NETMHC_PAN_1_20C is set. Specifies from which
length of peptide predictor works. Parameter value is number, with default 8. |
NETMHCPAN_1_20C_LENGTH_TO |
This variable has meaning only if WORK_NETMHC_PAN_1_20C is set. Specifies to which
length of peptide predictor works. Parameter value is number, with default 11. |
WORK_NETMHCPAN_1_24A |
Specifies execution of netMHCPan predictor, version 2.4a. Parameter value is string with default value "no". Possible values are "yes"; or "no" |
NETMHCPAN_1_24A_ALLELE_FILE |
This variable has meaning only if WORK_NETMHCPAN_1_24A is set. Specifies file with
allele. Path is specified absolute or relative with regard to configuration file. Parameter
value is string with default value "NetMhcPan.2.4a.pseudo".
|
NETMHCPAN_1_24A_LENGTH_FROM |
This variable has meaning only if WORK_NETMHCPAN_1_24A is set. Specifies from which
length of peptide predictor works. Parameter value is number, with default 8. |
NETMHCPAN_1_24A_LENGTH_TO |
This variable has meaning only if WORK_NETMHCPAN_1_24A is set. Specifies to which
length of peptide predictor works. Parameter value is number, with default 11. |
WORK_NETMHCPAN_1_28A |
Specifies execution of netMHCPan predictor, version 2.8a. Parameter value is string with default value "no". Possible values are "yes"; or "no" |
NETMHCPAN_1_28A_ALLELE_FILE |
This variable has meaning only if WORK_NETMHCPAN_1_28A is set. Specifies file with
allele. Path is specified absolute or relative with regard to configuration file. Parameter
value is string with default value "NetMhcPan.2.8a.pseudo".
|
NETMHCPAN_1_28A_LENGTH_FROM |
This variable has meaning only if WORK_NETMHCPAN_1_28A is set. Specifies from which
length of peptide predictor works. Parameter value is number, with default 8. |
NETMHCPAN_1_28A_LENGTH_TO |
This variable has meaning only if WORK_NETMHCPAN_1_28A is set. Specifies to which
length of peptide predictor works. Parameter value is number, with default 11. |
WORK_NETMHCPAN_2_10B |
Specifies execution of netMHCIIPan predictor, version 1.0b. Parameter value is string with default value "no". Possible values are "yes"; or "no" |
NETMHCPAN_2_10B_ALLELE_FILE |
This variable has meaning only if WORK_NETMHCPAN_2_10B is set. Specifies file with
allele. Path is specified absolute or relative with regard to configuration file. Parameter
value is string with default value "NetMhcIIPan.1.0b.pseudo".
|
NETMHCPAN_2_10B_LENGTH_FROM |
This variable has meaning only if WORK_NETMHCPAN_2_10B is set. Specifies from which
length of peptide predictor works. Parameter value is number, with default 9. |
NETMHCPAN_2_10B_LENGTH_TO |
This variable has meaning only if WORK_NETMHCPAN_2_10B is set. Specifies to which
length of peptide predictor works. Parameter value is number, with default 15. |
WORK_NETMHCPAN_2_20B |
Specifies execution of netMHCIIPan predictor, version 2.0b. Parameter value is string with default value "no". Possible values are "yes"; or "no" |
NETMHCPAN_2_20B_ALLELE_FILE |
This variable has meaning only if WORK_NETMHCPAN_2_20B is set. Specifies file with
allele. Path is specified absolute or relative with regard to configuration file. Parameter
value is string with default value "NetMhcIIPan.2.0b.pseudo".
|
NETMHCPAN_2_20B_LENGTH_FROM |
This variable has meaning only if WORK_NETMHCPAN_2_20B is set. Specifies from which
length of peptide predictor works. Parameter value is number, with default 9. |
NETMHCPAN_2_20B_LENGTH_TO |
This variable has meaning only if WORK_NETMHCPAN_2_20B is set. Specifies to which
length of peptide predictor works. Parameter value is number, with default 15. |
WORK_NETMHCPAN_2_30C |
Specifies execution of netMHCIIPan predictor, version 3.0c. Parameter value is string with default value "no". Possible values are "yes"; or "no" |
NETMHCPAN_2_30C_ALLELE_FILE |
This variable has meaning only if WORK_NETMHCPAN_2_30C is set. Specifies file with
allele. Path is specified absolute or relative with regard to configuration file. Parameter
value is string with default value "NetMhcIIPan.3.0c.pseudo". |
NETMHCPAN_2_30C_LENGTH_FROM |
This variable has meaning only if WORK_NETMHCPAN_2_30C is set. Specifies from which
length of peptide predictor works. Parameter value is number, with default 9. |
NETMHCPAN_2_30C_LENGTH_TO |
This variable has meaning only if WORK_NETMHCPAN_2_30C is set. Specifies to which
length of peptide predictor works. Parameter value is number, with default 15. |
1.4 Running
MassPred can be started with command in installed directory:
./work.sh <configuration_file> <input>
Input is a directory with FASTA files or a single FASTA file with multiple FASTA formats. For each protein in each FASTA file, MassPred executes required predictors and/or command if the command is given.
1.5 Results
After MassPred's successful ending, the result will be found in the output directory that is printed at
the end of execution.
The name of the output directory is calculated as the name of the input with added ".out".
If COMMAND variable is specified, in the output directory will be found the subdirectory command with the
results of the execution command for each protein. Other files are the results of execution requested predictor(s) on input data.
1.5.1 Generated files
The results of the predictor(s) application can be found in the output directory. Depending on which predictors are applied, the appropriate file with the results is generated. The files are compressed with gzip utility in order to save disk space and easier manipulation. The process always generate the LOAD files, while generating the SQL files is optional. The _fail files appear if the error occurred during the (any) predictor execution (the most common example is execution of VSL2b predictor which can not be applied on proteins with letters denotes ambiguous AAs).
Possible files in the output directory:
-
epitope_fail.load.gz
gziped LOAD file with list of netMHC failed predictors. This file is generated if the error occurred in the execution of netMHC predictors. -
epitope_fail.sql.gz
gziped SQL file with list of netMHC failed predictors. This file is generated if the error occurred in the execution of netMHC predictors. -
region_fail.load.gz
gziped LOAD file with list of disorder failed predictors. This file is generated if the error occurred in the execution of disorder predictors. -
region_fail.sql.gz
gziped SQL file with list of disorder failed predictors. This file is generated if the error occurred in the execution of disorder predictors. -
hydro.load.gz
gziped LOAD file with the results of hydrophobia. -
hydro.sql.gz
-
epitope_success.load.gz
gziped LOAD file with the results of netMHC predictors. -
epitope_success.sql.gz
gziped SQL file with the results of netMHC predictors. -
epitope_success_numeric.load.gz
gziped LOAD file with numeric results of netMHC predictors. This file is generated ifWORK_NUMERICoption is given. -
epitope_success_numeric.sql.gz
gziped SQL file with numeric results of netMHC predictors. This file is generated ifWORK_NUMERICoption is given. -
region_success.load.gz
gziped LOAD file with the results of disorder predictors. -
region_success.sql.gz
gziped SQL file with the results of disorder predictors. -
region_success_numeric.load.gz
gziped LOAD file with numeric results of disorder predictors. This file is generated ifWORK_NUMERICoption is given. -
region_success_numeric.sql.gz
results of disorder predictors. This file is generated ifWORK_NUMERICoption is given.
MassPred collects the results and filters them in a CSV file format in order to prepare the results in a form that can be used as an input in a load utility program for loading results in RDBMS tables. By default, the results are filtered for IBM DB2 RDBMS. Structure of the tables that can be used are:
epitope_fail ( protein_id VARCHAR(64), protein_reference VARCHAR(64), protein_file_name VARCHAR(64), type VARCHAR(32), allele VARCHAR(32), length INTEGER ) region_fail ( protein_id VARCHAR(64), protein_reference VARCHAR(64), protein_file_name VARCHAR(64), type VARCHAR(32) ) hydro ( protein_id VARCHAR(64), protein_reference VARCHAR(64), protein_file_name VARCHAR(64), position INTEGER, aa CHAR(1), hydro_kd DECIMAL, hydro_hw DECIMAL ) epitope ( protein_id VARCHAR(64), protein_reference VARCHAR(64), protein_file_name VARCHAR(64), position INTEGER, epitope VARCHAR(32), pos INTEGER, core VARCHAR(32), aff_log DECIMAL, aff DECIMAL, rank DECIMAL, binding CHAR(2), type VARCHAR(32), allele VARCHAR(32), length INTEGER ) epitope_numeric ( protein_id VARCHAR(64), protein_reference VARCHAR(64), protein_file_name VARCHAR(64), position INTEGER, epitope VARCHAR(32), pos INTEGER, core VARCHAR(32), aff_log DECIMAL, aff DECIMAL, rank DECIMAL, binding CHAR(2), type VARCHAR(32), allele VARCHAR(32), length INTEGER ) region ( protein_id VARCHAR(64), protein_reference VARCHAR(64), protein_file_name VARCHAR(64), begin INTEGER, end INTEGER, order CHAR(1), type VARCHAR(32) ) region_numeric ( protein_id VARCHAR(64), protein_reference VARCHAR(64), protein_file_name VARCHAR(64), position INTEGER, aa CHAR(1), value DECIMAL, order CHAR(1), type VARCHAR(32) )
1.5.2 Command
If the option WORK_COMMAND is set, then in subdirectory command, for each protein from input
FASTA files, three files with file name in form:
<name>.<number>.<suffix>, "name" is the name of input file and "number" is the position of the protein in FASTA file.
Meanning of the value for suffix are:
- "rc" - file with result code of executed command,
- "out" - file with standard output of executed command,
- "err" - file with standard error output of executed command.
The command gets from standard input content of FASTA file (which refers to the specific protein in
the order).
Before the execution of command, the next environment variables are set:
MASSPRED_INPUT- name of input FASTA file,MASSPRED_INPUT_POSITION- position of protein in input FASTA file,MASSPRED_OUTPUT- prefix of file name output files,MASSPRED_FASTA_DB- first field from header of input FASTA file,MASSPRED_FASTA_ID- second field from header of input FASTA file,MASSPRED_FASTA_REFERENCE_DB- third field from header of input FASTA file,MASSPRED_FASTA_REFERENCE- fourth field from header of input FASTA file,MASSPRED_FILE_NAME- base part of input FASTA file name.
1.6 Tests
Calling MassPred is very simple. On the command line from directory where MassPred is unpacked (for example in directory /usr/local/masspred), just execute work.sh script:
./work.sh my_directory/configuration.ish my_directory/my_file.faa
Where configuration.ish is configuration file described in chapter 1.3, and my_file.faa is
file that contains FASTA format of protein(s) or the directory with files containing FASTA protein code in FASTA format.
For example, the following commands can be used for the test of successful installation of MassPred:
./work.sh test/configuration.ish test/iedb_90149017.faa
./work.sh test/configuration.ish test/iedb_9000.faa
./work.sh test/configuration.ish test/dir
./work.sh test/configuration.ish test/cancer.faa
./work.sh test/configuration.ish test/p03211.faa
./work.sh test/configuration.ish test/NC_000913.faa
1.7 Uninstallation
For uninstalling MassPred just remove the directory in which MassPred is installed.
1.8 Update
The longer and more secure way is to uninstall MassPred first and then install it again.
The shorter way allows retain installed predictors. Before removing the directory containing the MassPred, move the directory
predictors (with installed predictors) to a temporary location. Remove the Masspred installation directory, and after that unpack
again MassPred (in the same directory) and replace directory predictors with the previous version (from the temporary location).
It is important that the absolute path to the predictors directory is not changed. In order to ensure
that, it is essential to always install the MassPred in the same directory (for example
/usr/local/masspred).
1.9 Possible problems
When the execution is normal, the MassPred cleans all the temporary directories, but in the case of execution terminates abnormally, it is possible that some temporary directories will not be properly removed. In this case, it is necessary to manually remove all temporary directories, which have form:
/tmp/masspreddir <number> <number>
When the execution is normal, the MassPred cleans all the temporary directories, but in the case of execution terminates abnormally, it is possible that some temporary directories will not be properly removed.
1.10 Application example
MassPred, is a script oriented system. It is started by running the script work.sh with two input parameters: name of the MassPred application configuration file and the name of the fasta file or directory which includes fasta files. For example,
/usr/local/masspred/work.sh configuration.ish p03211.faa
The configuration file (configuration.ish) includes parameters related to the current computer system (for example, the number of concurrent CPUs), and parameters related to supported predictors. An example of a configuration file is:
CPU_NUMBER=8
WORK_HYDRO=yes
WORK_ISUNSTRUCT=yes
WORK_VSL2=yes
WORK_NETMHC_1_34A=yes
NETMHC_1_34A_ALLELE_FILE=NetMhc.3.4a.pseudo
NETMHC_1_34A_LENGTH_FROM=8
NETMHC_1_34A_LENGTH_TO=9
...
File p03211.faa includes the fasta version of protein(s) on which the predictors are to be applied. The file can include one or more proteins in fasta format. For example,
>gi|119110|sp|P03211.1|EBNA1_EBVB9 RecName: Full=Epstein-Barr nuclear antigen 1; Short=EBNA-1; Short=EBV nuclear antigen 1 MSDEGPGTGPGNGLGEKGDTSGPEGSGGSGPQRRGGDNHGRGRGRGRGRGGGRPGAPGGSGSGPRHRDGV RRPQKRPSCIGCKGTHGGTGAGAGAGGAGAGGAGAGGGAGAGGGAGGAGGAGGAGAGGGAGAGGGAGGAG GAGAGGGAGAGGGAGGAGAGGGAGGAGGAGAGGGAGAGGGAGGAGAGGGAGGAGGAGAGGGAGAGGAGGA GGAGAGGAGAGGGAGGAGGAGAGGAGAGGAGAGGAGAGGAGGAGAGGAGGAGAGGAGGAGAGGGAGGAGA GGGAGGAGAGGAGGAGAGGAGGAGAGGAGGAGAGGGAGAGGAGAGGGGRGRGGSGGRGRGGSGGRGRGGS GGRRGRGRERARGGSRERARGRGRGRGEKRPRSPSSQSSSSGSPPRRPPPGRRPFFHPVGEADYFEYHQE GGPDGEPDVPPGAIEQGPADDPGEGPSTGPRGQGDGGRRKKGGWFGKHRGQGGSNPKFENIAEGLRALLA RSHVERTTDEGTWVAGVFVYGGSKTSLYNLRRGTALAIPQCRLTPLSRLPFGMAPGPGPQPGPLRESIVC YFMVFLQTHIFAEVLKDAIKDLVMTKPAPTCNIRVTVCSFDDGVDLPPWFPPMVEGAAAEGDDGDDGDEG GDGDEGEEGQE >gi|89902357|ref|YP_524828.1| 50S ribosomal protein L7/L12 [Rhodoferax ferrireducens T118] MAFDKDAFLTALDSMTVMELNDLVKAIEEKFGVSAAAMSAPAAGGAVAAVAEEKTEFNVVLLEAGAAKVS VIKAVREITGLGLKEAKDMVDGAPKNVKEGVSKVDAEAALKKLLDAGAKAELK .............
After execution, Masspred creates a directory with the added suffix .out to the name of the input file. The directory contains gzipped files with results for every required prediction. For example, for the part of the configuration file shown above, a directory p03211.faa.out was created with the following files:
epitope_success.load.gz- gzipped file with the predicted data from the epitope predictorshydro.load.gz- gzipped file with calculated hydrophobicity values for every AAregion_success.load.gz- gzipped file with the predicted data from the disorder predictors
All files are in TAB separated format and prepared for loading into RDBMS. For example, the content of region_success.load file is:
119110 P03211.1 p03211 1 413 D IsUnstruct 119110 P03211.1 p03211 414 414 O IsUnstruct 119110 P03211.1 p03211 415 480 D IsUnstruct 119110 P03211.1 p03211 481 541 O IsUnstruct 119110 P03211.1 p03211 542 554 D IsUnstruct 119110 P03211.1 p03211 555 613 O IsUnstruct 119110 P03211.1 p03211 614 641 D IsUnstruct 89902357 YP_524828.1 p03211 1 55 D IsUnstruct 89902357 YP_524828.1 p03211 56 84 O IsUnstruct 89902357 YP_524828.1 p03211 85 123 D IsUnstruct 119110 P03211.1 p03211 1 479 D VSL2b 119110 P03211.1 p03211 480 497 O VSL2b 119110 P03211.1 p03211 498 498 D VSL2b 119110 P03211.1 p03211 499 541 O VSL2b 119110 P03211.1 p03211 542 553 D VSL2b 119110 P03211.1 p03211 554 606 O VSL2b 119110 P03211.1 p03211 607 641 D VSL2b 89902357 YP_524828.1 p03211 1 5 D VSL2b 89902357 YP_524828.1 p03211 6 36 O VSL2b 89902357 YP_524828.1 p03211 37 44 D VSL2b 89902357 YP_524828.1 p03211 45 84 O VSL2b 89902357 YP_524828.1 p03211 85 123 D VSL2b
II. EpDis
EpDis is a tool for simultaneous access and visualization of different prediction methods, and combining and comparing results obtained by their application to the same input data, as well as calculation of consensus from results of different methods. It is developed to facilitate research for biologists and immunologists. EpDis can process protein sequences from different sources: FASTA files, RDBMS, Uniprot database or raw sequences. It supports execution of various T-Cell epitope predictors, disorder and disordered-binding predictors and methods for hydropathy calculation. EpDis tool is modular and configurable, easily extensible with additional predictors, implemented in Java.
Current version is 4.0. Download link EpDis.4.0.tar.gz
2.1 Requirements
In order to install and use EpDis application Oracle Java should be installed in the system, at least JRE 6. Java Development Kit (JDK) can be installed in the following way:
- download latest JDK distribution from http://www.oracle.com/technetwork/java/javase/downloads/index.html for your Linux distribution, as tar.gz archive and unpack it in some dedicated folder,
-
go to folder
/usr/binand create symbolic link with following command:sudo ln -s /home/Java/jdk1.6.0_29/bin/java javawhere
"/home/Java/jdk1.6.0_29"is path to the folder where JDK distribution was unpacked.
2.2 Installation
To install application execute following command:
java –jar epdis-install.jar
and follow these steps:
Step 1. - Welcome screen
First installation screen contains basic information about application. Click "Next".
Step 2. - Installation path selection
Select folder where application should be installed and click "Next". If there is no such folder on file system following message will be displayed:
Step 3. - Installation progress
Step 4. - Shortcuts
Step 5. - Installation completion
Remark: For further notice let %EPDIS_HOME% be the path to installation folder, as it was specified in Step 2. (in this example /home/daca/EPDIS).
2.3 Uninstallation
To uninstall application, go to folder %EPDIS_HOME%/Uninstaller/ and execute following command:
java –jar uninstaller.jar
2.4 Configuration
After instalation, there are customizable configuration files in %EPDIS_HOME%/config/ folder , which can be changed and accommodated for local environment:
epdis.properties
Contains global application configuration with following properties:
| Name | Description |
|---|---|
DataManagementModule.enabled |
Whether data management module is enabled or not. By default it is disabled. Enabling this module it is possible to load proteins from database and execute prediction methods on it, as well as saving of prediction results directly into the database through MassPred component. Possible values are true/false. |
ExecutionModule.show_not_configured_methods_on_startup |
Whether notification about not properly configured methods should be shown on application startup or not. (see next paragraph). Default value is true. Possible values are true/false |
MassPredModule.enabled |
Whether the MassPred is enabled or not. |
methods.properties
Contains properties of each prediction method. EpDis application comes with support for following methods:
For each method, the set of its properties is defined in the following way:
| Name | Value | Description |
|---|---|---|
netMhcPanPredictor.name |
NetMhcPan |
The name of the prediction method. Must be unique. |
netMhcPanPredictor.programHome |
/home/daca/dev/netMHCpan-2.4 |
Path to the method installation on local file system. *Normally this is the only value that should be changed. |
netMhcPanPredictor.programCommand |
${netMhcPanPredictor.programHome}/netMHCpan |
Program execution command. Usually should not be changed. Value of the expression ${netMhcPanPredictor.programHome} represents value of netMhcPanPredictor.programHome property. |
netMhcPanPredictor.allelesSource |
${netMhcPanPredictor.programHome}/data/MHC_pseudo.dat |
Path to the file with Allele&s definition. Usually should not be changed |
Usually, only value of netMhcPanPredictor.programHome should be changed to fit local environment.
During startup, application will check this configuration file, and load only those methods which are configured properly, i.e. it will load method if the path to the corresponding program installation is valid.
For those methods which are not configured properly, application will notify user if value of the property ExecutionModule.show_not_configured_methods_on_startup is set to true.
jdbc.properties
Contains properties related to database connectivity:
| Name | Description |
|---|---|
db.driverClass |
JDBC driver class, should not be changed |
db.port |
Database port |
db.host |
Database host |
db.name |
Database name, containing table definitions for application purposes |
db.default_schema |
Database schema |
db.url |
JDBC url, composed of values of other properties, should not be changed |
db.username |
User name |
db.password |
Password |
masspred.properties
| Name | Description |
|---|---|
massPred.Home |
Path to the method installation on local file system |
massPred.Command=./work.sh |
Program execution command. Should not be changed. |
2.5 Running
Running of EpDis can be performed in two following ways:
- from command line, executing
java -jar epdis-4.0.0.jarcommand from installation folder, or - by clicking on the shortcut icon from the start menu, created during installation (Step 4.)
2.5.1 Making predictions
After successful startup the following screen will appear:
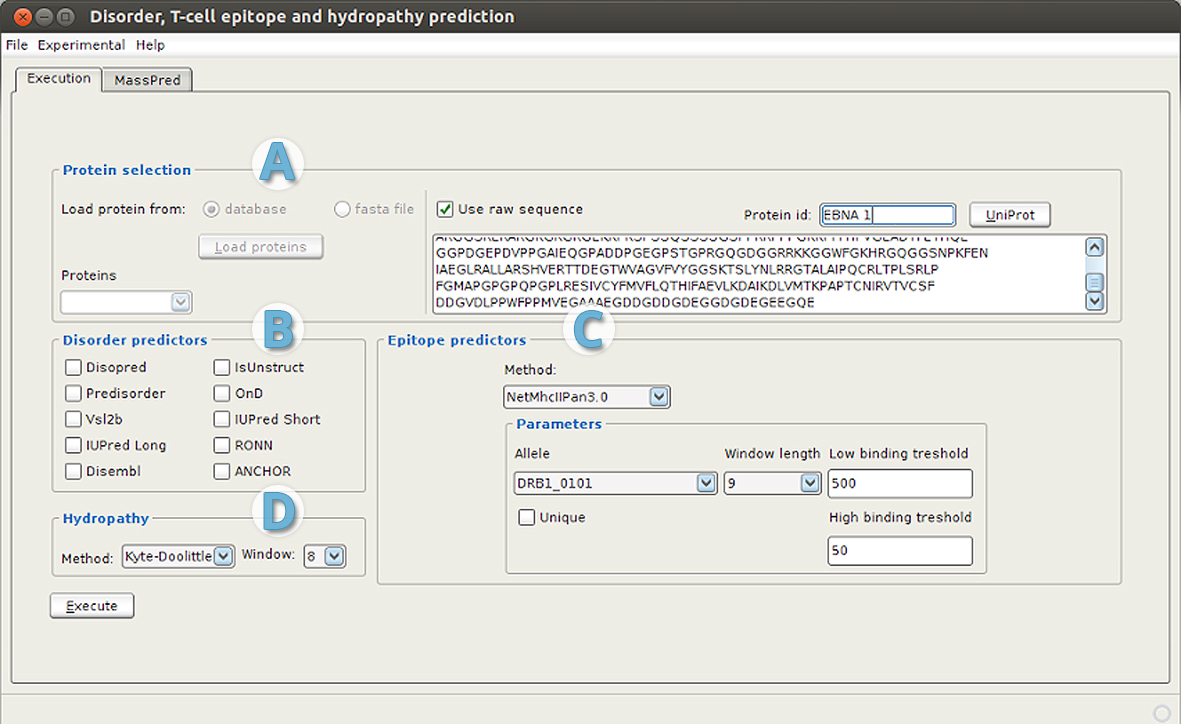
User interface is divided into tabs (one or two based on configuration). 'Execution' tab is composed of four parts:
- Protein selection (part A):
- from database
- from file in FASTA format
- from UNIPROT database, by specifying protein identifier
- as raw protein sequence
- Disorder and disorder-binding predictor selection (part B) - it is possible to choose one or more predictors to make predictions.
- Epitope predictor selection (part C) - single selection of different prediction method. In this example the NetMHCIIPan3.0 predictor is selected, and specified parameters: allele DRB1_0101, peptide of length 9 and default treshold. For MHC binding predictors results are values that represent the binding affinity of the peptides. The choice of an appropriate threshold is a critical task and should be chosen carefully. Inclusion of an experimental determined data about T cell epitopes and MHC binders might help in choosing an appropriate threshold value (see bellow).
- Hydropathy calculation (part D) - selection of hydropathy scale and peptide length.
It is possible to include in the display, beside predictors results, experimental data about secondary structures and T-cell epitopes. Entering experimental data is based on the addition of an appropriate xml file whose structure is in accordance with the pre-defined XML scheme:
Adding, deleting and overviewing of already added experimental data can be done by choosing corresponding action from 'Experimental' menu item. EpDis displays added experimental data in a form of table:
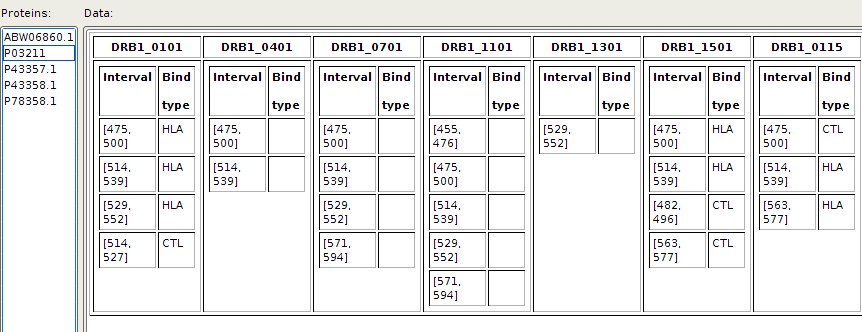
Predictions for selected set of predictors are done by clicking on the button 'Execute'. The panel with visualized results is presented:
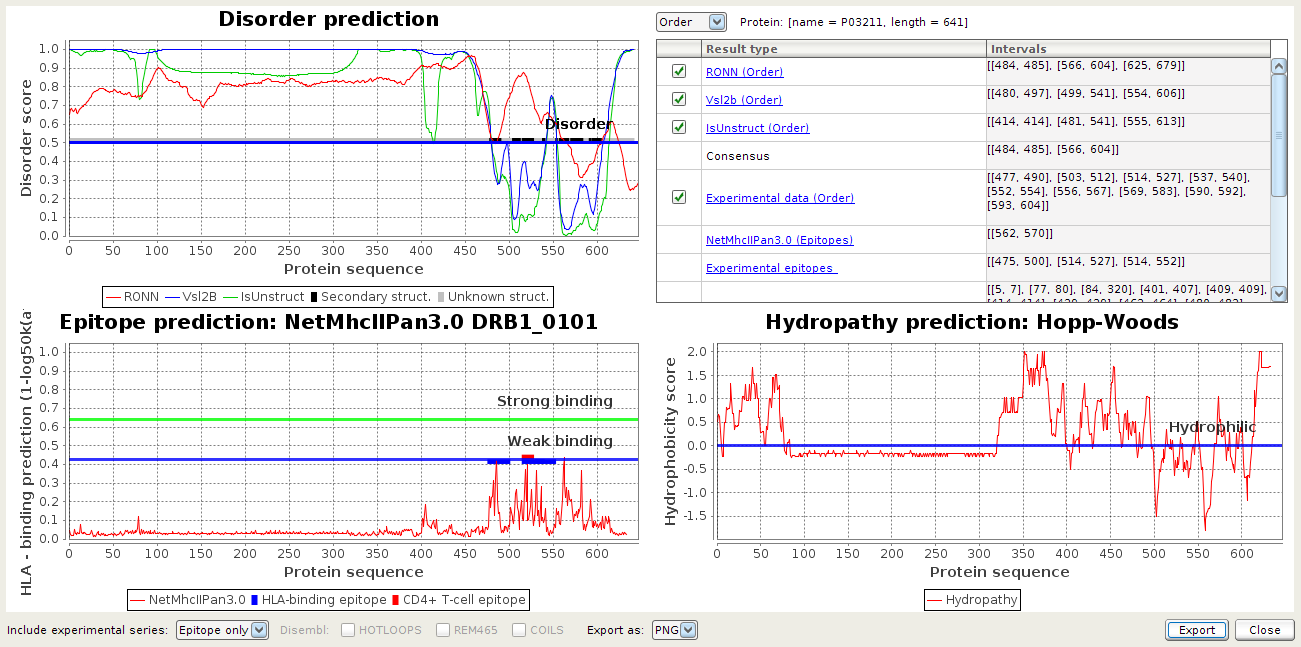
All the colors and annotations can be adjusted either in the configuration files or during runtime. It is possible to choose what is to be presented, whether intervals of ordered or of disordered parts of a protein. Each displayed disorder predictor result may be excluded/included from the graphic display (by checking/unchecking the check box next to the name of predictor in the top right table). The native result in the text form of each predictor can be displayed by clicking on predictor name. Each graphic can be individually exported to various graphic formats, such as PNG, PDF, SVG, and EPS. The experimentally validated T cell epitopes and MHC binders can be displayed in all three graphics or only 'epitope part D'. All the experimental data is displayed in in the actual length of the epitope and in the actual position within protein sequence.
2.5.2 MassPred integration
MassPred is fully integrated into EpDis tool through 'MassPred module'. This module enables the running of the MassPred tool from graphic mode. Turning on this component is optional and configurable. Apart from extending the functionalities of EpDis, the main purpose of this module is to facilitate the use of the MassPred tool itself, by allowing the user to specify its parameters simply by selecting the target predictors. The MassPred module has a predefined default configuration, but it allows storing and loading of a custom configuration as well. After execution of MassPred, the output is redirected and displayed to the user in the separate window. The interface for the MassPred module is shown in the following picture:
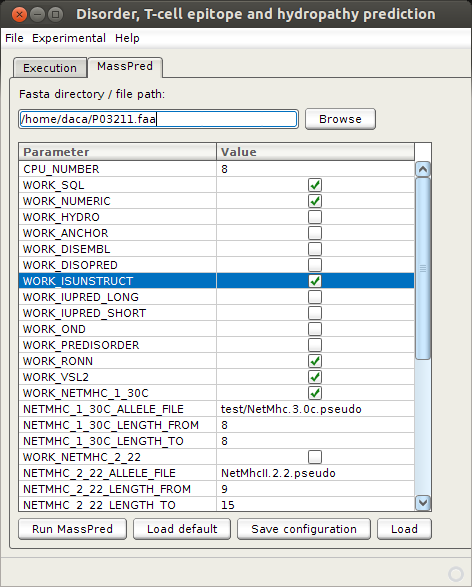
'WORK_SQL' in order to store prediction results into database; 'WORK_NUMERIC' - in order to import all results not just intervals od disorder/order, T cell epitopes, disorder-binding regions.
Specified disorder predictors are: 'WORK_ISUNSTRUCT', 'WORK_RONN', 'WORK_VSL2'.
Specified MHC binding predictors are: 'WORK_NETMHC1_30C' for peptide lenght form 'NETMHC1_30C_LENGTH_FROM' to 'NETMHC1_30C_LENGTH_to', in this example that are peptides of lenght 8 only, and specified HLA allele set from file test/NetMhc.3.0c.pseudo.
2.6 Database tables design
Apart from before mentioned tables needed for storing data from output of MassPred, following DDL should be executed to create all EpDis-related tables:
CREATE TABLE EPDIS_CACHE ( KEY VARCHAR(70) NOT NULL, PREDICTION_RESULT BLOB(1048576) INLINE LENGTH 164 NOT NULL, PRIMARY KEY (KEY) ); CREATE TABLE PROTEIN ( ID VARCHAR(20) NOT NULL, SEQUENCE VARCHAR(10000) NOT NULL, LENGTH SMALLINT NOT NULL, FASTA_HEADER VARCHAR(500) NOT NULL, ORD_NUM_IN_DISPROT VARCHAR(20) NOT NULL, PRIMARY KEY (ID) );
ER diagram describing MassPred/EpDis entities is given in the following image:

If 'DatabaseModule' is enabled tables 'EPITOPE_NUMERIC' and 'REGION_NUMERIC' are used as first level cache during predictors execution.
Table 'EPDIS_CACHE' is used as the second level cache.
III. Known issues
-
I have problem running 'IEDB ann' predictor
If exception is thrown during run of 'IEDB ann' predictor, with message that temporary dir could not be created, here's what needs to be done:- Open
NetMhc3.4/netMhcfile for editing - Change line
wwwrun = os.environ.get("USER") == Nonetowwwrun = False - Check if problem is solved
- Open
-
For all other issues, please send an e-mail with detailed steps needed for error reproduction.
Also it would be helpful to attach 'log' file, placed within
%EPDIS_HOME%/targetfolder.